Importance
Production of these proteins provides materials for experiments measuring the kinetics of self-assembly with techniques such as Surface Plasmon Resonance (SPR), solution scattering experiments using Small Angle X-ray Scattering (SAXS) and imaging using Transmission Electron Microscopy (TEM) and Total Internal Reflection Fluorescence (TIRF). Characterisation of these proteins is essential, as it ensures that any data obtained from these experiments is valid. It is also essential that the proteins are monodisperse, that is, of a single species, to ensure the quality and accuracy of data from experiments such as SPR and SAXS.
Aims
To produce, purify and characterise HIV-1 CA proteins, with and without mutations that:
- have disulfide bond cross-links to form discrete hexameric and pentameric structures;
- avoid dimerisation between CA proteins from adjacent hexamers;
- contain sites for reversible and irreversible conjugation to DNA.
Method Overview

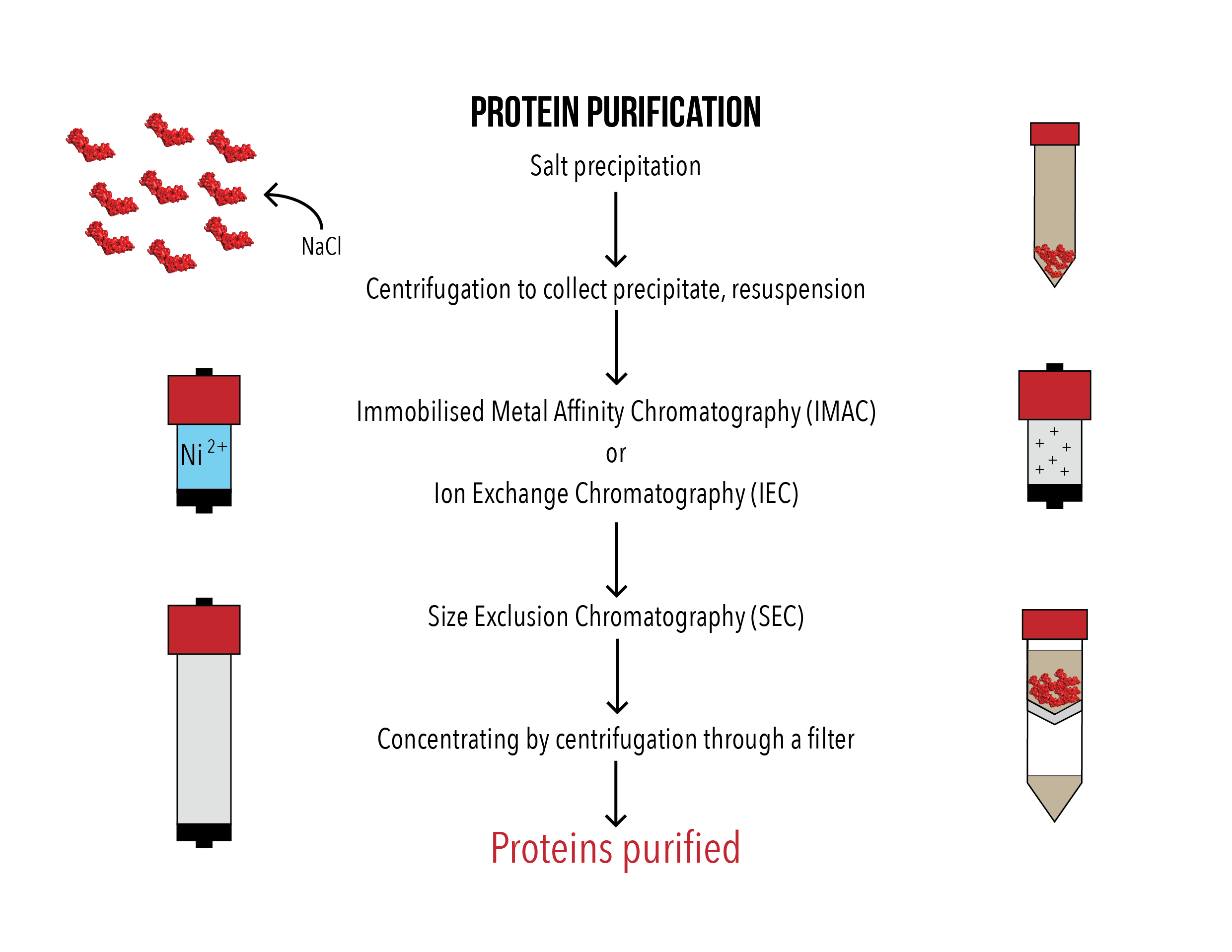
Figures 1 (left) and 2 (right): Flowcharts of CA protein production and purification method.
Plasmids containing the desired insert were heat shock transformed into Escherichia coli BL21 (DE3) pLysS cells. The bacteria were grown in Luria broth (LB) media at 37°C, induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to 0.4 mM when the optical density at 600 nm of the media reached 0.6 and then grown overnight at 20°C. The cell pellet was collected by centrifugation and lysed by sonication. The cell lysate was then centrifuged to remove cell debris, and only the soluble supernatant was collected.
NaCl was added to 2.5 M to precipitate the CA protein, as the CA protein aggregates under high salt conditions. After incubation on a spinning rotor at 4C overnight, the protein was collected by centrifugation, resuspended and a second salt precipitation was performed. After centrifugation and resuspension in a buffer with NaCl, the protein was purified with either Immobilised Metal Affinity Chromatography (IMAC) or Ion Exchange Chromatography (IEC) and Size Exclusion Chromatography (SEC). The purified protein was then concentrated by centrifugation using a 10 kDa filter and snap frozen in 10% glycerol.
Production
Heat shock transformation into chemically competent E. coli BL21DE3 (pLysS) cells
2 µL of plasmid construct was incubated with 18 µL of chemically competent E. coli BL21DE3 (pLysS) cells on ice for 20 minutes. The cells were then heat shocked for 45 seconds at 42C and placed back onto ice for 2 minutes. The cells were allowed to grow for 45 minutes in 200 µL of SOC outgrowth media (NEB) at 37°C and 170 rpm, spread plated onto Luria broth (LB) agar plates containing 100 µg/mL of ampicillin and 34 µg/mL of chloramphenicol and grown at 37°C overnight.
Generation of starter culture
One colony was selected from the plate grown overnight at random and grown in 15 mL of LB containing 100 µg/mL of ampicillin and 34 µg/mL of chloramphenicol at 37°C and 170 rpm until the optical density at 600 nm (OD600) > 1.
Large scale grow up of E. coli (4-8 L) at 37°C 160 rpm.
4 to 8 flasks containing 500 mL of LB containing 100 µg/mL of ampicillin and 34 µg/mL of chloramphenicol at 37°C were inoculated with the starter culture to a starting OD600 of 0.005. The cells were grown at 37°C and 170 rpm and OD600 was periodically measured. When the OD600 reached above 0.6, IPTG was added to a final concentration of 0.4 mM to induce the expression of the CA protein. After induction, the cells were grown overnight at 20°C and 170 rpm.
Collection of cells by centrifugation
The bacterial culture was centrifuged at 3340 x g for 45 minutes, and the cell pellet was collected and resuspended in 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide, 2mM DTT, 1 mg/ml lysozyme, a tip dip DNAse and 1 EDTA-protease inhibitor tablet.
Cell lysis by sonication, removal of cell debris
The cell pellet was lysed by sonication (Branson) for 6 minutes at 50% amplitude at alternating 1 second intervals, 15 seconds on and 15 seconds off, kept on ice. The cell lysate was ultra-centrifuged at 43150 x g for 30 minutes. The supernatant was collected and centrifuged again at 43150 x g for 30 minutes to remove any residual cell debris. The supernatant (soluble fraction) was collected.
Precipitation of the protein by incubation with NaCl
NaCl was added to the soluble fraction to 2.5 M and incubated on a spinning rotor at 4C overnight1. Protein precipitate was collected by centrifugation at 4000 x g for 45 minutes at 4C, and resuspended in 15 mL of 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide, 2 mM DTT and 1 protease inhibitor tablet. The protein was precipitated again by repeating the salt precipitation process to try to purify the protein further before chromatography.
SDS-PAGE gel
Samples were collected at various stages in the protein production process, and examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 180 V for 25 minutes. The samples included: bacterial culture before induction, bacterial culture after induction, cell lysate, soluble fraction, first NaCl precipitation supernatant, second NaCl precipitation supernatant and resuspended protein after salt precipitation. NuPAGE LDS loading dye (ThermoFisher), prepared with final concentration of 1 mM DTT, was added to each sample before loading onto precast SDS-PAGE gel.
Purification
Two stages of fast protein liquid chromatography (FPLC) were performed for protein purification. Samples were run on an AKTApurifier liquid chromatography pump (GE Healthcare).
IMAC
Immobilised metal ion affinity chromatography (IMAC) was performed to purify mutant CA proteins containing a polyhistidine motif2. His-tagged protein was bound to a 5 mL HisTrap FF Crude column (GE Healthcare) in buffer containing 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide and 10 mM imidazole, and eluted with buffer containing 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide and 500 mM imidazole. Fractions were collected and examined with SDS-PAGE. Eluted proteins were concentrated with an Amicon Centrifugal Filter Unit with Ultracel-10 Membrane (Merck) to above 2 mg/mL if the eluted fractions were of a lower concentration.
As an alternative to using the AKTApurifier liquid chromatography pump, a gravity column containing Profinity IMAC Ni-charged Resin (Biorad) was used for IMAC. This enabled the elution of protein in a smaller volume. The Ni-charged resin was washed with water 3 times, then equilibrated in 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide and 10 mM imidazole. Salt purified proteins were incubated with the resin overnight at 4C, then loaded onto a gravity column. Proteins were eluted with buffer containing 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide and 500 mM imidazole. Fractions were collected and examined with SDS-PAGE. Eluted proteins were concentrated with an Amicon Centrifugal Filter Unit with Ultracel-10 Membrane (Merck) to above 2 mg/mL if the eluted fractions were of a lower concentration.
IEC
Mutant CA proteins without a polyhistidine motif were purified by anion exchange chromatography (IEC) instead of IMAC. CA protein was loaded onto a 5 mL HiTrap Q HP Column equilibrated in 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide, 2 mM DTT, and fractions were collected as the sample was loaded. Fractions were collected and examined with SDS-PAGE. Eluted proteins were concentrated with an Amicon Centrifugal Filter Unit with Ultracel-10 Membrane (Merck) to above 2 mg/mL if the eluted fractions were of a lower concentration.
SEC
Following IMAC or IEC, the concentrated eluted proteins were purified further by size exclusion chromatography (SEC). 5 mL of concentrated eluted mutant CA proteins were loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) and eluted with buffer containing 50 mM Tris-HCl (pH 8.0), 0.02% NaAzide and 2 mM DTT over 1.2 column volumes. Any collected fractions containing eluted proteins were examined by SDS-PAGE. Eluted proteins were concentrated with an Amicon Centrifugal Filter Unit with Ultracel-10 Membrane (Merck).
Long term storage
Glycerol was added to purified protein a final concentration of 10%. The proteins were then snap frozen in liquid nitrogen and stored at -80C.
Results
Each protein solution was run through two stages of purification, firstly, either ion exchange chromatography (IEC) or ion exchange metal chromatography (IMAC), and secondly size exclusion chromatography (SEC). At each stage, fractions corresponding to the largest elution peaks were collected, and run on an SDS-PAGE gel. Figure 3 shows presence of clear bands at the correct molecular weight for each protein, confirming their purification.
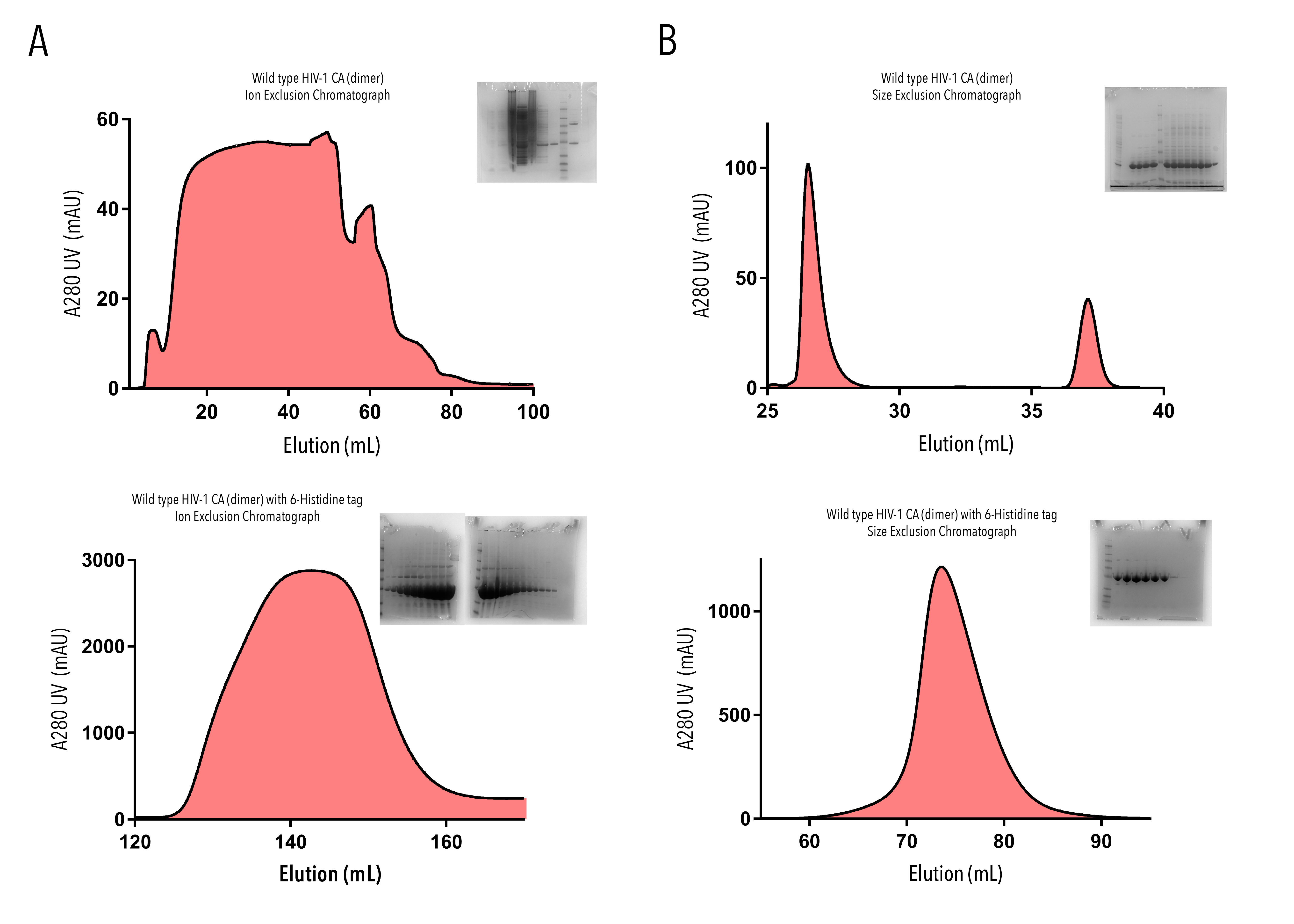
Figure 3: Purification of HIV-1 CA proteins with A280 UV absorbance elution profiles and SDS-gel confirmation, from (A) Ion Exchange Chromatography and (B) Size Exclusion Chromatograhy. Flow through of Wild type HIV-1 CA (dimer) (top row) and Wild-type HIV-1 CA (dimer) with 6-Histidine tag (bottom row) were collected in 1-2mL fractions. Fractions corresponding to the largest elution peaks were run on SDS-PAGE gels.
Due to technical difficulties with our AKTA machine, we were unable to perform IMAC on some of our proteins with this method. An alternative nickel gravity flow column was used to separate out our target proteins based upon their affinity to nickel ions. Results of these exclusions post-SEC are shown in Figure 4. Fractions corresponding to the largest peaks on these elutions were run on SDS-PAGE gel to confirm purification of our target protein. Bands of the correct molecular weight were observed across proteins.
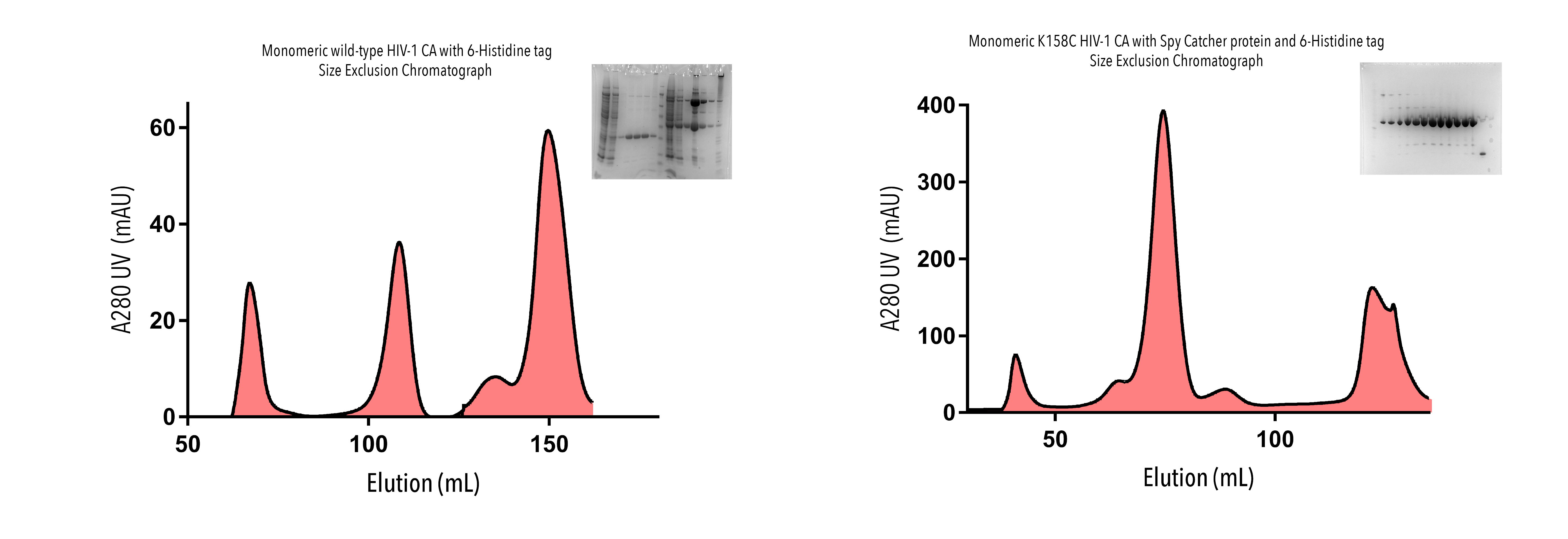
Figure 4: Purification of HIV-CA proteins with A280 UV absorbance elution profiles and SDS-gel confirmation, from Size Exclusion Chromatography. Flow-through of monomeric wild-type HIV-1 CA with 6-Histidine tag (top left), K158C HIV-1 CA (dimer) with Spy Catcher and 6-Histidine tag (top right), and monomeric K158C HIV-1 CA with Spy Catcher and 6-Histidine tag (bottom) were collected in 1-2mL fractions. Fractions corresponding to the largest elution peaks were run on SDS-PAGE gels.
Conclusion
We have successfully produced and purified a number of HIV-1 CA protein mutants, each with different protein binding capabilities for use in analytical experiments.
Large scale production and purification has been confirmed for the following constructs:
- Monomeric wild-type HIV-1 CA with 6-Histidine tag
- This protein can be conjugated reversibly to our DNA origami scaffold, for use in kinetic experiments measuring the inter-hexamer protein binding events.
- Wild type HIV-1 CA (dimer)
- As the native protein for found in vivo, this protein serves as a positive control for analytical experiments.
- Wild-type HIV-1 CA (dimer) with 6-Histidine tag
- This protein can be conjugated reversibly to our DNA origami scaffold, for use in kinetic experiments measuring properties of the native HIV-1 CA proteins.
- Monomeric K158C HIV-1 CA with Spy Catcher3 and 6-Histidine tag
- This protein can be conjugated both reversibly (via. 6-Histidine tag) and irreversibly (via. Spy Catcher protein) to our DNA origami scaffold for use in kinetic experiments. It also is able to be fluorescently tagged (via. Accessible cysteine) for use in single molecule imaging.
Small scale production and purification has been confirmed for the following constructs:
- K158C (dimer) HIV-1 CA
- This protein can be irreversibly conjugated to our DNA origami scaffold for use in analytical experiments. It can also be conjugated to a fluorophore for single molecule analysis.
- K158C (dimer) HIV-1 CA with 6-Histidine tag
- This protein can be reversibly conjugated to our DNA origami scaffold (via. 6-Histidine) for use in analytical experiments. It can also simultaneously be conjugated to a fluorophore (via accessible cysteine) for single molecule analysis.
- Cross-linked pentamer HIV-1 CA
- This pentameric protein can be used to observe intra-pentamer or hexamer interactions. It may be included in experiments attempting lattice formation of HIV-1 CA proteins.
- Discrete cross-linked pentamer HIV-1 CA with 6-Histidine tag
- This protein may be used to determine the solution structure of a formed HIV-1 CA pentamer subunit. It may also be used to determine affinity of formed pentameric proteins to single stranded DNA.
- Discrete cross-linked hexamer HIV-1 CA with 6-Histidine tag
- This protein may be used to determine the solution structure of a formed HIV-1 CA hexamer subunit with a polyhistidine tag. It has also been used to determine affinity of formed hexameric proteins to single stranded DNA.
By combining information about isolated protein binding events, enabled by each of these HIV-1 CA mutants, we are able to piece together a larger story about what fundamental principles drive the assembly of the HIV-1 viral capsid in vivo.
In the future we would like to express and purify all of our successfully cloned constructs. In addition to production of target proteins, we have developed a robust protocol for purification of the HIV-1 CA protein and its variants. This procedure has been modified to specifically suit properties of the HIV-1 CA protein, and ensuring purity from other proteins produced during expression, and isolation monodisperse protein solutions.
References
- Ehrlich, L. S., Agresta B. E. & Carter C. A. (1992) Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J Virol 66(8), pp. 4874-83.
- Hochuli, E., Bannwarth, W., Döbeli, H., Gentz, R. and Stüber, D. (1988). Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent. Nature Biotechnology, 6(11), pp.1321-1325.
- Li, L., Fierer, J., Rapoport, T. and Howarth, M. (2014). Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag. J Mol Biol, 426(2), pp.309-317.